An interatomic potential, traditionally regarded as a mathematical function, serves to depict atomic interactions within molecules or solids by expressing potential energy concerning atom positions. These potentials are pivotal in materials science and engineering, facilitating atomic-scale simulations, predictive material behavior, accelerated discovery, and property optimization. Notably, the landscape is evolving with machine learning transcending conventional mathematical models. Various machine learning-based interatomic potentials, such as artificial neural networks, kernel-based methods, deep learning, and physics-informed models, have emerged, each wielding unique strengths and limitations. These methods decode the intricate connection between atomic configurations and potential energies, offering advantages like precision, adaptability, insights, and seamless integration. The transformative potential of machine learning-based interatomic potentials looms large in materials science and engineering. They promise tailor-made materials discovery and optimized properties for specific applications. Yet, formidable challenges persist, encompassing data quality, computational demands, transferability, interpretability, and robustness. Tackling these hurdles is imperative for nurturing accurate, efficient, and dependable machine learning-based interatomic potentials primed for widespread adoption in materials science and engineering. This roadmap offers an appraisal of the current machine learning-based interatomic potential landscape, delineates the associated challenges, and envisages how progress in this domain can empower atomic-scale modeling of the composition-processing-microstructure-property relationship, underscoring its significance in materials science and engineering.
Purpose-led Publishing is a coalition of three not-for-profit publishers in the field of physical sciences: AIP Publishing, the American Physical Society and IOP Publishing.
Together, as publishers that will always put purpose above profit, we have defined a set of industry standards that underpin high-quality, ethical scholarly communications.
We are proudly declaring that science is our only shareholder.
ISSN: 1361-651X
Serving the multidisciplinary materials community, the journal aims to publish new research work that advances the understanding and prediction of material behaviour at scales from atomistic to macroscopic through modelling and simulation.
Yong-Wei Zhang et al 2025 Modelling Simul. Mater. Sci. Eng. 33 023301
Filip Šiška et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055022
The Inconel IN939 superalloy is a suitable material for additive manufacturing. However, such a manufacturing process creates an anisotropic microstructure, which influences the material’s mechanical performance. The effect of microstructure on fatigue behavior at high temperatures is studied using the finite element method within the crystal plasticity framework. The spatial evolution of stresses and strains with cyclic loading is analyzed. The results show that material loaded perpendicularly to the building direction (BD) has higher strengthening and higher activity of slip systems in whole volume while loading parallel to the BD induces plasticity, preferably in grains with (101) and (111) orientation. Such differences can lead to different damage initiation and evolution.
V Román et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055013
Proton exchange membrane fuel cells face performance limitations owing to their high electrical contact resistance (ECR) at the gas diffusion layer (GDL)-bipolar plate interface. In this study, a stochastic random Poisson process is employed to numerically generate GDL realizations coupled with various machine learning (ML) techniques, such as deep neural networks (DNNs) and random forests (RFs), which permit the identification of those descriptors characterizing a nonwoven carbon paper GDL that predict the pressure dependence of ECR when compressing the GDL. The DNN trained on a representative number of GDL realizations is used to accurately predict ECR for various sets of descriptors and RF is used to identify the importance of these descriptors within the sets. Using the so-classified descriptors, the ultimate goal is to reduce ECR occurring at the clamping pressure by manufacturing the GDL accordingly. Obviously, compression is shown to be the most important for predicting the pressure dependence of ECR however, some of the descriptors considered here, like fiber length, fiber density, bond density, and fiber-to-bond ratio, seem to be more crucial than traditional and widely used ones, such as porosity and the orientation of fibers, for example. These unexpected findings are just the first results of a large-scale simulation campaign, supported by ML techniques to reduce computational cost, which will presumably identify the GDL manufacturing parameters that unambiguously determine the ECR.
Alexander Stukowski 2010 Modelling Simul. Mater. Sci. Eng. 18 015012
The Open Visualization Tool (OVITO) is a new 3D visualization software designed for post-processing atomistic data obtained from molecular dynamics or Monte Carlo simulations. Unique analysis, editing and animations functions are integrated into its easy-to-use graphical user interface. The software is written in object-oriented C++, controllable via Python scripts and easily extendable through a plug-in interface. It is distributed as open-source software and can be downloaded from the website http://ovito.sourceforge.net/.
Stefan Bauer et al 2024 Modelling Simul. Mater. Sci. Eng. 32 063301
Science is and always has been based on data, but the terms ‘data-centric’ and the ‘4th paradigm’ of materials research indicate a radical change in how information is retrieved, handled and research is performed. It signifies a transformative shift towards managing vast data collections, digital repositories, and innovative data analytics methods. The integration of artificial intelligence and its subset machine learning, has become pivotal in addressing all these challenges. This Roadmap on Data-Centric Materials Science explores fundamental concepts and methodologies, illustrating diverse applications in electronic-structure theory, soft matter theory, microstructure research, and experimental techniques like photoemission, atom probe tomography, and electron microscopy. While the roadmap delves into specific areas within the broad interdisciplinary field of materials science, the provided examples elucidate key concepts applicable to a wider range of topics. The discussed instances offer insights into addressing the multifaceted challenges encountered in contemporary materials research.
Thomas Rocke and James R Kermode 2025 Modelling Simul. Mater. Sci. Eng. 33 055020
The problem of constructing a dataset for MLIP development which gives the maximum quality in the minimum amount of compute time is complex, and can be approached in a number of ways. We introduce a ‘Bayesian selection’ approach for selecting from a candidate set of structures, and compare the effectiveness of this method against other common approaches in the task of constructing ideal datasets targeting Silicon surface energies. We show that the Bayesian selection method performs much better than Simple Random Sampling at this task (for example, the error on the (100) surface energy is 4.3× lower in the low data regime), and is competitive with a variety of existing selection methods, using ACE (Drautz 2019 Phys. Rev. B 99 014104) and MACE (Batatia et al 2022 Advances in Neural Information Processing Systems pp 11423–36) features.
Erik van der Giessen et al 2020 Modelling Simul. Mater. Sci. Eng. 28 043001
Modeling and simulation is transforming modern materials science, becoming an important tool for the discovery of new materials and material phenomena, for gaining insight into the processes that govern materials behavior, and, increasingly, for quantitative predictions that can be used as part of a design tool in full partnership with experimental synthesis and characterization. Modeling and simulation is the essential bridge from good science to good engineering, spanning from fundamental understanding of materials behavior to deliberate design of new materials technologies leveraging new properties and processes. This Roadmap presents a broad overview of the extensive impact computational modeling has had in materials science in the past few decades, and offers focused perspectives on where the path forward lies as this rapidly expanding field evolves to meet the challenges of the next few decades. The Roadmap offers perspectives on advances within disciplines as diverse as phase field methods to model mesoscale behavior and molecular dynamics methods to deduce the fundamental atomic-scale dynamical processes governing materials response, to the challenges involved in the interdisciplinary research that tackles complex materials problems where the governing phenomena span different scales of materials behavior requiring multiscale approaches. The shift from understanding fundamental materials behavior to development of quantitative approaches to explain and predict experimental observations requires advances in the methods and practice in simulations for reproducibility and reliability, and interacting with a computational ecosystem that integrates new theory development, innovative applications, and an increasingly integrated software and computational infrastructure that takes advantage of the increasingly powerful computational methods and computing hardware.
Modesar Shakoor 2025 Modelling Simul. Mater. Sci. Eng. 33 053001
The level-set (LS) method has been widely spread since its introduction in 1988. One of its main features is that interfaces are represented through signed distance functions, and the distance property eases the smoothing of discontinuities across an interface, and the computation of normal vectors and mean curvatures. This property can be lost whenever the LS function is evolved, for instance, through an LS transport equation. Numerous studies proposing so-called LS reinitialization methods to restore the distance property have been published since 1988. This paper is a review of numerical developments on LS reinitialization in the past decade (2014–2024). LS reinitialization methods are classified into three categories: direct methods which geometrically compute distances, local indirect methods which solve the Eikonal equation point-by-point, and global indirect methods which solve the Eikonal equation for all points at once. The review focuses on numerical methods and investigates the following questions. Can it be implemented in a parallel computing environment with nearly optimal scalability? Can it be used with any approximation method, and is it compatible with unstructured grids? Can it be extended to reach higher-order convergence rates? Can it be combined with mass change error attenuation techniques? Does it involve any numerical parameters that may affect its robustness? Is it limited only to some applications? Through a quantitative and qualitative analysis of the past decade’s literature, this review paper proposes novel insights on LS reinitialization. Research on direct methods should focus on parallel efficiency and robust higher-order distance computation techniques. More attention should be given to local indirect methods, especially regarding parallel and higher-order algorithms for unstructured grids. Control parameters for global indirect methods should be better determined or eliminated. More research is needed on the issue of blind spots in two-phase problems involving moving contact lines and their elimination.
Tesia D Janicki et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055010
Stilbenes are a class of organic compounds with broad-ranging pharmaceutical and agricultural applications, which are typically isolated and purified through recrystallization. We are motivated by reducing experimental waste and optimizing yield via developing predictive simulations for processing-dependent crystal morphologies. Using resveratrol as a model stilbene system, we have developed an approach for simulating crystallization with molecular resolution using on-lattice kinetic Monte Carlo. In this work, we highlight modifications to the Stochastic Parallel PARticle Kinetic Simulator (SPPARKS) software package, which were essential to this application. Key enhancements include the incorporation of non-orthogonal cell shapes and monomer anisotropy approximations using bound hard spheres. This new SPPARKS application has been applied to resveratrol with attachment energy libraries obtained from density functional theory, resulting in excellent agreement with experimental morphology prediction.
Ian Wise et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055011
Cross slip is a dislocation mechanism that significantly impacts the mechanical behavior of engineering alloys. Here, we advance a 3D phase-field dislocation dynamics (PFDD) mesoscale technique to simulate cross slip across a broad range of face-centered cubic (FCC) metals. The formulation incorporates elastic anisotropy, an FCC numerical grid, and a high-fidelity representation of the entire γ-surface from density functional theory for eight FCC metals and no adjustable parameters or rules. The relaxed core structures under zero stress for all metals are predicted to extend in plane. The analytical model for stacking fault width agrees well with the PFDD result under the assumption of elastic isotropy but overestimates it under elastic anisotropy, when the degree of anisotropy is large. The dynamic simulations are designed to elucidate the material parameters that influence the propensity for cross slip. Whether cross slip occurs under a non-Schmid stress or to bypass a hard obstacle, the critical stress to cross slip scales strongly with the anisotropic energy coefficient for a screw dislocation.
Sindu B S and Jan Hamaekers 2025 Modelling Simul. Mater. Sci. Eng. 33 065010
The conventional way of developing epoxy polymers may not be fully efficient as it is constrained by fixed compositions that limit performance. To overcome this, we introduce a machine learning (ML)-based approach that accurately predicts mechanical properties from its basic structural features, enabling broader design exploration. The results from molecular dynamics simulations have been used to derive the ML model. The salient feature of our work is that for the development of epoxy polymers based on EPON-862, several new hardeners were explored in addition to the conventionally used ones. The influence of additional parameters like the proportion of curing agent used and the extent of curing on the mechanical properties of epoxy polymers were also investigated. This method can be further extended by providing the epoxy polymer with the desired properties through knowledge of the structural characteristics of its constituents. The findings of our study can thus lead toward development of efficient design methodologies for epoxy polymeric systems.
Ali Reza Safi et al 2025 Modelling Simul. Mater. Sci. Eng. 33 065009
In this study, the role of elastic and interfacial energies in the shape evolution of T1 precipitates in Al–Cu–Li alloys is investigated using phase-field modeling. We employ a formulation considering the stoichiometric nature of the precipitate phase explicitly, including coupled equation systems for various order parameters. Inputs such as elastic properties are derived from density functional theory calculations, while chemical potentials are obtained from CALPHAD databases. This methodology provides a framework that is consistent with the derived chemical potentials to study the interplay of thermodynamic, kinetic, and elastic effects on T1 precipitate evolution in Al–Cu–Li alloys. It is shown that diffusion-controlled lengthening and interface-controlled thickening are important mechanisms to describe the growth of T1 precipitates. Furthermore, the study illustrates that the precipitate shape is significantly influenced by the anisotropy in interfacial energy and linear reaction rate, however, elastic effects are only of secondary importance.
Zhong Zhang et al 2025 Modelling Simul. Mater. Sci. Eng. 33 065008
Piezoelectric materials are essential in various fields due to their unique electromechanical coupling effect. However, the computational efficiency and accuracy of these materials across multiple physical domains require enhancement. Here, the hygro-mechanical-electrical (HME) coupling isogeometric analysis method (HMEC-IGA) is proposed to explore the mechanical response of piezoelectric structures in hygro environments. Based on the B-spline theory, the basic equations of piezoelectric materials and boundary conditions, the non-uniform rational B-spline basis function is introduced to construct displacement and electric potential shape functions, and nodes are interpolated to determine the displacement of each point in the solution domain. Using the principle of virtual work and the Newmark method, the control equation and motion equation of HMEC-IGA were derived. Numerical examples show HMEC-IGA achieves sufficient precision with fewer control points or units, and is more effective than the finite element method. HMEC-IGA holds significant potential for application in the design, development, and performance analysis of piezoelectric materials.
Zhe-Zhi Jiang and Jia-Lin Tsai 2025 Modelling Simul. Mater. Sci. Eng. 33 065007
This study adopted an experimental approach to evaluate the capability of the constitutive model proposed in literature, in conjunction with two different failure criteria, for predicting the tensile strength of fiber metal laminates (FMLs). The FMLs containing layers of fiber composites and thin aluminum sheets with the stacking sequences of [Al/(0/90)4]S [Al/(0/90/±45)2]S [Al/(0/±30/60)2]S and [Al/(±30/±60)2]S. were fabricated. Quasi-static tensile tests were conducted on the samples, from which the failure stresses were obtained. A constitutive model of FMLs including the nonlinear behaviors of composites together with failure criterion was employed to predict the tensile strength of FMLs. Maximum stress criterion and Hill–Tsai criterion were considered as failure criterion in the fiber composites. The thermal residual stresses generated during curing process within each layer were calculated using laminated plate theory and employed in the analysis. Results demonstrated that for FMLs containing 0° carbon fiber plies, maximum stress criterion provided tensile strength predictions that closely matched the experimental data. Conversely, for FMLs composed exclusively of off-axis plies, Hill–Tsai criterion yielded more accurate predictions. Furthermore, the influence of nonlinear effects of fiber composites varied depending on the laminate configuration. For FMLs with 0° carbon fiber plies, the nonlinear behavior had minimal impact on the predicted tensile strength. However, for FMLs without 0° plies, nonlinear effects played a significant role in tensile strength predictions.
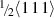 vacancy and interstitial prismatic dislocation loops in tungsten
vacancy and interstitial prismatic dislocation loops in tungstenJan Fikar et al 2025 Modelling Simul. Mater. Sci. Eng. 33 065006
The vacancies and interstitials produced in high-energy collision cascades of irradiated tungsten can form prismatic dislocation loops with Burgers vectors  and
and  . The
. The  loops are very mobile, and their mobility is essential for the microstructure development of irradiated materials, It is a key parameter for predictive models such as kinetic Monte Carlo. We investigated the mobility of
loops are very mobile, and their mobility is essential for the microstructure development of irradiated materials, It is a key parameter for predictive models such as kinetic Monte Carlo. We investigated the mobility of  vacancy and interstitial hexagonal loops as a function of their size using the recent embedded-atom method tungsten potential. The phonon drag phenomenon occurs at high temperatures and can be separated during post-processing from the thermally activated motion. The magnitude of the phonon drag at 300 K was evaluated and appeared to be critical for single interstitial atoms, with a nearly ten-fold increase of their diffusion, while dislocation loops are less influenced.
vacancy and interstitial hexagonal loops as a function of their size using the recent embedded-atom method tungsten potential. The phonon drag phenomenon occurs at high temperatures and can be separated during post-processing from the thermally activated motion. The magnitude of the phonon drag at 300 K was evaluated and appeared to be critical for single interstitial atoms, with a nearly ten-fold increase of their diffusion, while dislocation loops are less influenced.
Modesar Shakoor 2025 Modelling Simul. Mater. Sci. Eng. 33 053001
The level-set (LS) method has been widely spread since its introduction in 1988. One of its main features is that interfaces are represented through signed distance functions, and the distance property eases the smoothing of discontinuities across an interface, and the computation of normal vectors and mean curvatures. This property can be lost whenever the LS function is evolved, for instance, through an LS transport equation. Numerous studies proposing so-called LS reinitialization methods to restore the distance property have been published since 1988. This paper is a review of numerical developments on LS reinitialization in the past decade (2014–2024). LS reinitialization methods are classified into three categories: direct methods which geometrically compute distances, local indirect methods which solve the Eikonal equation point-by-point, and global indirect methods which solve the Eikonal equation for all points at once. The review focuses on numerical methods and investigates the following questions. Can it be implemented in a parallel computing environment with nearly optimal scalability? Can it be used with any approximation method, and is it compatible with unstructured grids? Can it be extended to reach higher-order convergence rates? Can it be combined with mass change error attenuation techniques? Does it involve any numerical parameters that may affect its robustness? Is it limited only to some applications? Through a quantitative and qualitative analysis of the past decade’s literature, this review paper proposes novel insights on LS reinitialization. Research on direct methods should focus on parallel efficiency and robust higher-order distance computation techniques. More attention should be given to local indirect methods, especially regarding parallel and higher-order algorithms for unstructured grids. Control parameters for global indirect methods should be better determined or eliminated. More research is needed on the issue of blind spots in two-phase problems involving moving contact lines and their elimination.
Yong-Wei Zhang et al 2025 Modelling Simul. Mater. Sci. Eng. 33 023301
An interatomic potential, traditionally regarded as a mathematical function, serves to depict atomic interactions within molecules or solids by expressing potential energy concerning atom positions. These potentials are pivotal in materials science and engineering, facilitating atomic-scale simulations, predictive material behavior, accelerated discovery, and property optimization. Notably, the landscape is evolving with machine learning transcending conventional mathematical models. Various machine learning-based interatomic potentials, such as artificial neural networks, kernel-based methods, deep learning, and physics-informed models, have emerged, each wielding unique strengths and limitations. These methods decode the intricate connection between atomic configurations and potential energies, offering advantages like precision, adaptability, insights, and seamless integration. The transformative potential of machine learning-based interatomic potentials looms large in materials science and engineering. They promise tailor-made materials discovery and optimized properties for specific applications. Yet, formidable challenges persist, encompassing data quality, computational demands, transferability, interpretability, and robustness. Tackling these hurdles is imperative for nurturing accurate, efficient, and dependable machine learning-based interatomic potentials primed for widespread adoption in materials science and engineering. This roadmap offers an appraisal of the current machine learning-based interatomic potential landscape, delineates the associated challenges, and envisages how progress in this domain can empower atomic-scale modeling of the composition-processing-microstructure-property relationship, underscoring its significance in materials science and engineering.
Stefan Bauer et al 2024 Modelling Simul. Mater. Sci. Eng. 32 063301
Science is and always has been based on data, but the terms ‘data-centric’ and the ‘4th paradigm’ of materials research indicate a radical change in how information is retrieved, handled and research is performed. It signifies a transformative shift towards managing vast data collections, digital repositories, and innovative data analytics methods. The integration of artificial intelligence and its subset machine learning, has become pivotal in addressing all these challenges. This Roadmap on Data-Centric Materials Science explores fundamental concepts and methodologies, illustrating diverse applications in electronic-structure theory, soft matter theory, microstructure research, and experimental techniques like photoemission, atom probe tomography, and electron microscopy. While the roadmap delves into specific areas within the broad interdisciplinary field of materials science, the provided examples elucidate key concepts applicable to a wider range of topics. The discussed instances offer insights into addressing the multifaceted challenges encountered in contemporary materials research.
David Furrer 2023 Modelling Simul. Mater. Sci. Eng. 31 073001
Materials and manufacturing engineering are continuing to advance in part to computational materials and process modeling and associated linkages with associated interdisciplinary efforts across all engineering, manufacturing, and quality disciplines. Computational modeling has enabled virtual processing, prediction and assessment of potential new materials and manufacturing processes, without or with limited need to perform costly and time-consuming physical trials. Development and integration of computational materials and process engineering requires a number of seemingly disparate critical technical elements, making this evolving computational capability very complicated. Accurate and validated models are supporting rapid material, process, and component development, and additionally qualification and certification of new final products through integrated computational materials engineering (ICME). These capabilities are driving further industrial utilization of computational material and process modeling with formalized linkages and integration within multidisciplinary engineering workflows. Past utilization, present applications and potential future development activities indicate that industry has now fully embraced the tools and methods, and overarching engineering framework of ICME.
Vikram Gavini et al 2023 Modelling Simul. Mater. Sci. Eng. 31 063301
Electronic structure calculations have been instrumental in providing many important insights into a range of physical and chemical properties of various molecular and solid-state systems. Their importance to various fields, including materials science, chemical sciences, computational chemistry, and device physics, is underscored by the large fraction of available public supercomputing resources devoted to these calculations. As we enter the exascale era, exciting new opportunities to increase simulation numbers, sizes, and accuracies present themselves. In order to realize these promises, the community of electronic structure software developers will however first have to tackle a number of challenges pertaining to the efficient use of new architectures that will rely heavily on massive parallelism and hardware accelerators. This roadmap provides a broad overview of the state-of-the-art in electronic structure calculations and of the various new directions being pursued by the community. It covers 14 electronic structure codes, presenting their current status, their development priorities over the next five years, and their plans towards tackling the challenges and leveraging the opportunities presented by the advent of exascale computing.
Luo et al
This study employs molecular dynamics simulations to investigate the structural characteristics and surface tension behaviors of Fe-C and Fe-N melts at various compositions and temperatures. Through analysis of atomic distributions, radial distribution functions, and coordination numbers, we reveal key relationships between atomic behavior and surface phenomena. Our findings indicate that the enrichment of C and N at the melt surface, along with the contraction of Fe atoms into the liquid phase, results in density and pressure gradients that influence surface tension. Increased C and N concentrations enhance surface enrichment and intensify Fe contraction. Notably, the first peak heights of radial distribution functions for Fe-C and C-C pairs, as well as Fe-N and N-N pairs, show opposing trends with rising concentrations, reflecting competition between these atomic species. In the Fe-C system, surface tension steadily decreases, while the Fe-N system experiences an initial increase in surface tension before peaking at 0.2 wt% N and then declining. As temperature increases, surface tension decreases, accompanied by a weakening of short-range order. These insights enhance our understanding of the impact of C and N enrichment on Fe-based melts.
Lu et al
Single-crystal silicon, an anisotropic material, exhibits distinct differences in cutting characteristics across various crystal orientations. This paper studies the influence of elliptical vibration cutting (EVC) on the anisotropic cutting behavior of single-crystal silicon through molecular dynamics simulation. The elliptical vibration nano-cutting is performed along two typical crystallographic directions of each of the (100), (110), and (111) crystal planes. The findings reveal considerable anisotropy in the surface consistency, material removal rate, stress, phase transformation, cutting force, and cutting temperature of single-crystal silicon when cut across different crystal orientations. Specifically, the [0-11] and [010] crystallographic directions on the (100) plane demonstrate the highest surface integrity, while the [1-10] direction on the (110) plane shows the lowest. In terms of material removal rate, the [00-1] direction on the (110) plane has the greatest rate (79.1%), whereas the [-1-12] direction on the (111) plane records the smallest (70.8%). Analysis of the coordination number and radial distribution function reveals less phase changes in the [0-11], [010] directions on the (100) plane and the [00-1] direction on the (110) plane. Notably, the [00-1] direction on the (110) plane contains about 27% fewer 5-coordinated atoms than the [0-11] direction on the (111) plane. The cutting force analysis indicates that the mean tangential forces in the (100) [0-11], (100) [010], and (110)[00-1] directions are smaller, suggesting a relatively easier cutting process in these directions.
Yu et al
The melting behavior of aluminum alloy is crucial for spacecraft structure re-entry damage. This work performs the molecular dynamics simulations to investigate the amorphization behavior of polycrystalline AlMg alloys with different grain sizes and pre-deformations under high temperatures. The simulation results demonstrated that the melting rate slows down for the polycrystalline AlMg alloys with the larger grains. The melting process shifts from the grain-boundary-to-interior amorphization in small grains to both grain boundary and interior melting in large grains. Further simulations discovered that the elastic pre-deformation has little effect on melting behaviors of polycrystalline AlMg alloys, and the plastic pre-deformation leads to the significant impact on the amorphization process of the smaller grain size but less contribution for the larger grains. This is originated from the fact that the small grains experience grain-boundary sliding and the large grains generate dislocations when the pre-plastic deformation. Analysis of the differences between the pre-loading and non-pre-loading cases reveals that the difference under plastic pre-loading is more obvious than elastic pre-loading. Such difference first rises then falls with increasing the temperature. In addition, the relationship between the difference in amorphous-atom volume fraction (pre-loading vs non-pre-loading) and grain size is further discussed and the pre-stretched (pre-compressed) deformation makes the difference decrease (increase) with grain size. The findings in this work could be helpful to understand the melting process of Al alloys under the high temperature environment.
ELBARNATY et al
Our work consists in modelling the effective properties of a piezoelectric-based nanocomposite that will be used for aeronautical applications. For this purpose, the PolyVinyliDene Fluoride (PVDF) is considered as a polymer piezoelectric matrix in which Graphene nanoRibbons (GnR) are spatially oriented as inclusions. Two different methods were used to estimate the effective properties: the Mori-Tanaka (MT) micromechanics scheme and the finite Element (FE) analysis. In the MT homogenization scheme, the electro-elastic Eshelby tensor was first computed for an anisotropic matrix. The entire MT scheme was implemented in the MATLAB environment. Afterwards a FE model of a nanocomposite unit cell was developed with ABAQUS in order to estimate the effective properties. The effective properties computed with the MT scheme showed good agreement with the predictions of the FE model. The orientation of GnR inclusions as well as their volume fraction in the PVDF matrix were found to significantly affect the effective electro-elastic properties of the GnR/PVDF nanocomposite. In addition, several physical mechanisms such as stacking-up and alignment defects of GnR were analyzed for their negative influence on the reinforcement effect of GnR. Our theoretical study will help in understanding the favorable effects of nano-inclusions over piezoelectric nanocomposites.
Leimeroth et al
Machine learning interatomic potentials (MLIPs) have massively changed the field of atomistic modeling. They enable the accuracy of density functional theory in large-scale simulations while being nearly as fast as classical interatomic potentials. Over the last few years, a wide range of different types of MLIPs have been developed, but it is often difficult to judge which approach is the best for a given problem setting. For the case of structurally and chemically complex solids, namely Al-Cu-Zr and Si-O, we benchmark a range of machine learning interatomic potential approaches, in particular, the Gaussian approximation potential (GAP), high-dimensional neural network potentials (HDNNP), moment tensor potentials (MTP), the atomic cluster expansion (ACE) in its linear and nonlinear version, neural equivariant interatomic potentials (NequIP), Allegro, and MACE. We find that nonlinear ACE and the equivariant, message-passing
graph neural networks NequIP and MACE form the Pareto front in the accuracy vs. computational cost trade-off. In case of the Al-Cu-Zr system we find that MACE and Allegro offer the highest accuracy, while NequIP outperforms them for Si-O. Furthermore, GPUs can massively accelerate the MLIPs, bringing them on par with and even ahead of non-accelerated classical interatomic potentials (IPs) with regards to accessible timescales. Finally, we explore the extrapolation behavior of the corresponding potentials, probe the smoothness of the potential energy surfaces, and finally estimate the user friendliness of the corresponding fitting codes and molecular dynamics interfaces.
Sindu B S and Jan Hamaekers 2025 Modelling Simul. Mater. Sci. Eng. 33 065010
The conventional way of developing epoxy polymers may not be fully efficient as it is constrained by fixed compositions that limit performance. To overcome this, we introduce a machine learning (ML)-based approach that accurately predicts mechanical properties from its basic structural features, enabling broader design exploration. The results from molecular dynamics simulations have been used to derive the ML model. The salient feature of our work is that for the development of epoxy polymers based on EPON-862, several new hardeners were explored in addition to the conventionally used ones. The influence of additional parameters like the proportion of curing agent used and the extent of curing on the mechanical properties of epoxy polymers were also investigated. This method can be further extended by providing the epoxy polymer with the desired properties through knowledge of the structural characteristics of its constituents. The findings of our study can thus lead toward development of efficient design methodologies for epoxy polymeric systems.
Ali Reza Safi et al 2025 Modelling Simul. Mater. Sci. Eng. 33 065009
In this study, the role of elastic and interfacial energies in the shape evolution of T1 precipitates in Al–Cu–Li alloys is investigated using phase-field modeling. We employ a formulation considering the stoichiometric nature of the precipitate phase explicitly, including coupled equation systems for various order parameters. Inputs such as elastic properties are derived from density functional theory calculations, while chemical potentials are obtained from CALPHAD databases. This methodology provides a framework that is consistent with the derived chemical potentials to study the interplay of thermodynamic, kinetic, and elastic effects on T1 precipitate evolution in Al–Cu–Li alloys. It is shown that diffusion-controlled lengthening and interface-controlled thickening are important mechanisms to describe the growth of T1 precipitates. Furthermore, the study illustrates that the precipitate shape is significantly influenced by the anisotropy in interfacial energy and linear reaction rate, however, elastic effects are only of secondary importance.
Salah ELBARNATY et al 2025 Modelling Simul. Mater. Sci. Eng.
Our work consists in modelling the effective properties of a piezoelectric-based nanocomposite that will be used for aeronautical applications. For this purpose, the PolyVinyliDene Fluoride (PVDF) is considered as a polymer piezoelectric matrix in which Graphene nanoRibbons (GnR) are spatially oriented as inclusions. Two different methods were used to estimate the effective properties: the Mori-Tanaka (MT) micromechanics scheme and the finite Element (FE) analysis. In the MT homogenization scheme, the electro-elastic Eshelby tensor was first computed for an anisotropic matrix. The entire MT scheme was implemented in the MATLAB environment. Afterwards a FE model of a nanocomposite unit cell was developed with ABAQUS in order to estimate the effective properties. The effective properties computed with the MT scheme showed good agreement with the predictions of the FE model. The orientation of GnR inclusions as well as their volume fraction in the PVDF matrix were found to significantly affect the effective electro-elastic properties of the GnR/PVDF nanocomposite. In addition, several physical mechanisms such as stacking-up and alignment defects of GnR were analyzed for their negative influence on the reinforcement effect of GnR. Our theoretical study will help in understanding the favorable effects of nano-inclusions over piezoelectric nanocomposites.
Zhe-Zhi Jiang and Jia-Lin Tsai 2025 Modelling Simul. Mater. Sci. Eng. 33 065007
This study adopted an experimental approach to evaluate the capability of the constitutive model proposed in literature, in conjunction with two different failure criteria, for predicting the tensile strength of fiber metal laminates (FMLs). The FMLs containing layers of fiber composites and thin aluminum sheets with the stacking sequences of [Al/(0/90)4]S [Al/(0/90/±45)2]S [Al/(0/±30/60)2]S and [Al/(±30/±60)2]S. were fabricated. Quasi-static tensile tests were conducted on the samples, from which the failure stresses were obtained. A constitutive model of FMLs including the nonlinear behaviors of composites together with failure criterion was employed to predict the tensile strength of FMLs. Maximum stress criterion and Hill–Tsai criterion were considered as failure criterion in the fiber composites. The thermal residual stresses generated during curing process within each layer were calculated using laminated plate theory and employed in the analysis. Results demonstrated that for FMLs containing 0° carbon fiber plies, maximum stress criterion provided tensile strength predictions that closely matched the experimental data. Conversely, for FMLs composed exclusively of off-axis plies, Hill–Tsai criterion yielded more accurate predictions. Furthermore, the influence of nonlinear effects of fiber composites varied depending on the laminate configuration. For FMLs with 0° carbon fiber plies, the nonlinear behavior had minimal impact on the predicted tensile strength. However, for FMLs without 0° plies, nonlinear effects played a significant role in tensile strength predictions.
Niklas Leimeroth et al 2025 Modelling Simul. Mater. Sci. Eng.
Machine learning interatomic potentials (MLIPs) have massively changed the field of atomistic modeling. They enable the accuracy of density functional theory in large-scale simulations while being nearly as fast as classical interatomic potentials. Over the last few years, a wide range of different types of MLIPs have been developed, but it is often difficult to judge which approach is the best for a given problem setting. For the case of structurally and chemically complex solids, namely Al-Cu-Zr and Si-O, we benchmark a range of machine learning interatomic potential approaches, in particular, the Gaussian approximation potential (GAP), high-dimensional neural network potentials (HDNNP), moment tensor potentials (MTP), the atomic cluster expansion (ACE) in its linear and nonlinear version, neural equivariant interatomic potentials (NequIP), Allegro, and MACE. We find that nonlinear ACE and the equivariant, message-passing
graph neural networks NequIP and MACE form the Pareto front in the accuracy vs. computational cost trade-off. In case of the Al-Cu-Zr system we find that MACE and Allegro offer the highest accuracy, while NequIP outperforms them for Si-O. Furthermore, GPUs can massively accelerate the MLIPs, bringing them on par with and even ahead of non-accelerated classical interatomic potentials (IPs) with regards to accessible timescales. Finally, we explore the extrapolation behavior of the corresponding potentials, probe the smoothness of the potential energy surfaces, and finally estimate the user friendliness of the corresponding fitting codes and molecular dynamics interfaces.
Filip Šiška et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055022
The Inconel IN939 superalloy is a suitable material for additive manufacturing. However, such a manufacturing process creates an anisotropic microstructure, which influences the material’s mechanical performance. The effect of microstructure on fatigue behavior at high temperatures is studied using the finite element method within the crystal plasticity framework. The spatial evolution of stresses and strains with cyclic loading is analyzed. The results show that material loaded perpendicularly to the building direction (BD) has higher strengthening and higher activity of slip systems in whole volume while loading parallel to the BD induces plasticity, preferably in grains with (101) and (111) orientation. Such differences can lead to different damage initiation and evolution.
Thomas Rocke and James R Kermode 2025 Modelling Simul. Mater. Sci. Eng. 33 055020
The problem of constructing a dataset for MLIP development which gives the maximum quality in the minimum amount of compute time is complex, and can be approached in a number of ways. We introduce a ‘Bayesian selection’ approach for selecting from a candidate set of structures, and compare the effectiveness of this method against other common approaches in the task of constructing ideal datasets targeting Silicon surface energies. We show that the Bayesian selection method performs much better than Simple Random Sampling at this task (for example, the error on the (100) surface energy is 4.3× lower in the low data regime), and is competitive with a variety of existing selection methods, using ACE (Drautz 2019 Phys. Rev. B99 014104) and MACE (Batatia et al 2022 Advances in Neural Information Processing Systems pp 11423–36) features.
Emilio N M Cirillo et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055014
Film formation from solvent evaporation in polymer ternary solutions is relevant for several technological applications, such as the fabrication of organic solar cells. The performance of the final device will strongly depend on the internal morphology of the obtained film, which, in turn, is affected by the processing conditions. We are interested in modeling morphology formation in 3D for ternary mixtures using both a lattice model and its continuous counterpart in the absence of evaporation. In our previous works, we found that, in 2D, both models predict the existence of two distinct regimes: (i) a low-solvent regime, characterized by two interpenetrated domains of the two polymers, and (ii) a high-solvent regime, where isolated polymer domains are dispersed in the solvent background. In the significantly more intriguing 3D case, we observe a comparable scenario both for the discrete and the continuous model. The lattice model reveals its ability to describe morphology formation even in the high solvent content 3D case, in which the three-dimensional nature of space could have prevented cluster formation. To realize the simulations we have written specific codes using the languages C and julia. The codes closely follows the algorithmic dynamics governing the lattice and the continuum model.
V Román et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055013
Proton exchange membrane fuel cells face performance limitations owing to their high electrical contact resistance (ECR) at the gas diffusion layer (GDL)-bipolar plate interface. In this study, a stochastic random Poisson process is employed to numerically generate GDL realizations coupled with various machine learning (ML) techniques, such as deep neural networks (DNNs) and random forests (RFs), which permit the identification of those descriptors characterizing a nonwoven carbon paper GDL that predict the pressure dependence of ECR when compressing the GDL. The DNN trained on a representative number of GDL realizations is used to accurately predict ECR for various sets of descriptors and RF is used to identify the importance of these descriptors within the sets. Using the so-classified descriptors, the ultimate goal is to reduce ECR occurring at the clamping pressure by manufacturing the GDL accordingly. Obviously, compression is shown to be the most important for predicting the pressure dependence of ECR however, some of the descriptors considered here, like fiber length, fiber density, bond density, and fiber-to-bond ratio, seem to be more crucial than traditional and widely used ones, such as porosity and the orientation of fibers, for example. These unexpected findings are just the first results of a large-scale simulation campaign, supported by ML techniques to reduce computational cost, which will presumably identify the GDL manufacturing parameters that unambiguously determine the ECR.
Ian Wise et al 2025 Modelling Simul. Mater. Sci. Eng. 33 055011
Cross slip is a dislocation mechanism that significantly impacts the mechanical behavior of engineering alloys. Here, we advance a 3D phase-field dislocation dynamics (PFDD) mesoscale technique to simulate cross slip across a broad range of face-centered cubic (FCC) metals. The formulation incorporates elastic anisotropy, an FCC numerical grid, and a high-fidelity representation of the entire γ-surface from density functional theory for eight FCC metals and no adjustable parameters or rules. The relaxed core structures under zero stress for all metals are predicted to extend in plane. The analytical model for stacking fault width agrees well with the PFDD result under the assumption of elastic isotropy but overestimates it under elastic anisotropy, when the degree of anisotropy is large. The dynamic simulations are designed to elucidate the material parameters that influence the propensity for cross slip. Whether cross slip occurs under a non-Schmid stress or to bypass a hard obstacle, the critical stress to cross slip scales strongly with the anisotropic energy coefficient for a screw dislocation.